Epidemiology and diagnostic criteria
Complications
Neurofibromatosis type 1 (NF1) is a genetic disease of autosomal dominant inheritance, linked to a mutation in the NF1 gene NF1 encoding neurofibromin. The genetics of neurofibromatosis is included in a dedicated chapter. Its prevalence is 1/4000 1–4 and its incidence estimated at one in 3000 births. 1,2,5–7Its phenotypic expression is variable even between members of a family. Its penetrance is complete at the age of 8 years: 97% of patients with NF1 meet the diagnostic criteria at 8 years 8.
The diagnosis is based on the combination of at least 2 of the 7 NIH criteria from 19889 revised in 202110, and summarized below. They differ depending on whether the patient has one of his two parents also affected.
REVISED DIAGNOSTIC CRITERIA FOR NF1
(1988 NIH CONSENSUS CONFERENCE, REVISED IN 2021)
A. The diagnostic criteria for NF1 are met in an individual who does not have a parent diagnosed with NF1 if two or more of the following are present:
- Six or more "café-au-lait" macules over 5 mm in greatest diameter in prepubertal individuals and over 15 mm in greatest diameter in post-pubertal individuals.
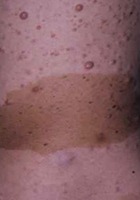
- Freckling in the axillary or inguinal region.* #
- Two or more neurofibromas of any type or one plexiform neurofibroma.
- Optic pathway glioma.
- Two or more iris Lisch nodules identified by slit lamp examination or two or more choroidal abnormalities (CAs)—defined as bright, patchy nodules imaged by optical coherence tomography (OCT)/near-infrared reflectance (NIR) imaging.
- A distinctive osseous lesion such as sphenoid dysplasia***, anterolateral bowing of the tibia, or pseudarthrosis of a long bone.
- A heterozygous pathogenic NF1NF1 variant with a variant allele fraction of 50% in apparently normal tissue such as white blood cells.
B. A child of a parent who meets the diagnostic criteria specified in A merits a diagnosis of NF1 if one or more of the criteria in A are present.
* : If only café-au-lait macules and freckling are present, the diagnosis is most likely NF1 but exceptionally the person might have another diagnosis such as Legius syndrome.
# : At least one of the two pigmentary findings (café-au-lait macules or freckling) should be bilateral.
** : Sphenoid wing dysplasia is not a separate criterion in case of an ipsilateral orbital plexiform neurofibroma.
-
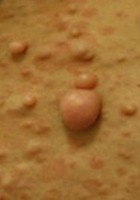
- Cutaneous neurofibromas (adult)
-
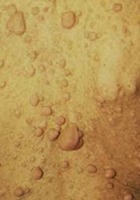
- Cutaneous neurofibromas (adult)
-
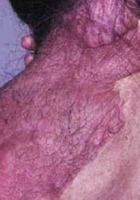
- Posterior cervical plexiform neurofibroma (adult)
-
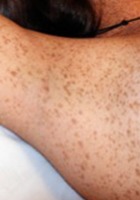
- Freckling in the axillary region (adult)
-
- Freckling in the axillary region (adult)
-
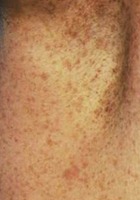
- "Café-au-lait" macules associated with cutaneous neurofibromas
-
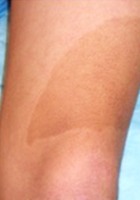
- Large "café-au-lait" macules (adult)
-
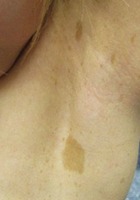
- "Café-au-lait" macules associated with freckling in the bilateral axillary region(child)
-
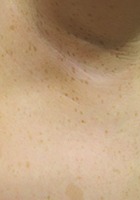
- "Café-au-lait" macules associated with freckling in the bilateral axillary region(child)
-
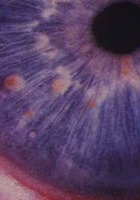
- Lisch nodules (slit lamp examination)
The disease complications are numerous, benign or malignant, somatic and psychological, and appear throughout the lifespan11. However, one of the peculiarities of NF1 remains its variability of expression from one patient to another and even within a family (where the genetic abnormality is identical). This makes it impossible to predict the clinical course of an affected child.
There are different types of neurofibromas: dermal (>95% of patients)12,13, peripheral nodular (at least 20% of patients)12and nodular orplexiform (20% of patients)14The main risk, inherent in nodular and plexiform neurofibromas, is the transformation into a malignant peripheral nerve sheath tumour (MPNST, formerly known as malignant nerve sheath tumour). The cumulative risk of developing MPNST during a patient's lifetime has been estimated to be 8-16%. It occurs mostly between 20 and 35 years of age. 15–19This risk is increased in patients with an NF1 phenotype, defined by the presence of subcutaneous neurofibromas: in case of subcutaneous neurofibromas, the risk of developing an internal plexiform neurofibroma or MPNST is multiplied by 3, and in case of plexiform neurofibroma, the risk of developing an MPNST is multiplied by 2020The tumour may be aggressive17, with a decreased life expectancy. Metabolic imaging such as PET-MRI or PET-scanner can diagnose a neurofibroma in the dysplastic stage and thus improve the prognosis through early surgery. Neurofibromas can also have a significant aesthetic and functional impact.
Optic pathway glioma 22–26 is a benign tumour with low malignancy potential present in 15-20% of children with NF1 22,27–31. On the other hand, its local extension can lead to exophthalmos, decreased visual acuity, or compression of the optic chiasma with possible repercussions on the gonadotropic hypothalamic-pituitary axis and early puberty 31,32.
Orthopaedic disorders are classic: congenital dysplasia of long bones (7.2% of patients) 33, sphenoidal dysplasia (1 to 7% of patients) 34 and scoliosis (10 to 28% of patients) 35. Congenital long bone dysplasia causes deformity, and an increased risk of fracture with pseudoarthrosis in 2 to 3.6% of patients 33,36Sphenoidal dysplasia can be secondary to plexiform neurofibroma 37, and can lead to pulsatile exophthalmos without decreased visual acuity and a hernia of the temporal lobe in the eyeball in case of total absence of sphenoid bone 38. Scoliosis, the most common orthopaedic manifestation, is often associated with vertebral dysplasia 39Patients with NF1 appear to be at increased risk of weakening osteopathy, with osteopenia presentin 48% of patients or osteoporosis in 25% 40,41.
Early stunting is present in one-third of children 42, precocious puberty can also occur 34,43. Precocious puberty is defined by the appearance of sexual criteria before the age of 7-8 years in girls and 9-10 years in boys.
Cardiovascular complications are mainly linked to essential or secondary hypertension, which can begin in childhood (16 to 19% of children with NF1) and whose risk increases with age 44–48. Secondary causes of high blood pressure associated with NF1 include: vascular nephropathy, paraganglioma, pheochromocytoma, or coarctation of the aorta. Congenital heart defects have also been reported in 0.4 to 8.6% of patients. 49–54There is also a significant risk of bleeding, particularly during neurofibroma surgery 55 ; risk attributed to vascular dysplasia and primary hemostasis abnormalities 56.
Epilepsy and its developmental repercussions are the most severe central neurological complication. Epilepsy affects 8% of patients 57,58, and is resistant to treatment in 30% of cases 59. In the latter cases, it can be associated with severe mental retardation.
Moderate cognitive impairment, outside of epilepsy, is the most common neurological complication 60, with an intelligence quotient in the lower middle part, behavioural disorders, and specific learning difficulties (visuospatial and visuomotor disorders, language disorders, fine and gross motor disorders, and executive function disorders)5,60–65. Other pathologies have also been associated with NF1, including attention deficit/hyperactivity, autistic, and psychosocial disorders 63,65.
Finally, patients with NF1 have an increased risk of neoplasia 66. Concerning the risk of breast cancer, particularly studied in the NF1 patient population, it occurs in women under 50 years of age 67–71 with a higher mortality rate 72. These characteristics justify the need of systematic annual screening by combining mammography-ultrasounds 73 et d’une IRM mammaire, à partir de l’âge de 30 ans chez les femmes atteintes de NF1. Les TMGN représentent 60% des cancers dans cette population mais les patients peuvent également développer d’autres tumeurs du spectre neurologique : les tumeurs cérébrales, dont les gliomes des voies optiques, mais aussi les gliomes du tronc cérébral 74, or gastrointestinal neuroendocrine tumours, which often occur at a younger age than that of the general population and that are often multiple and regularly located in the small intestine 75,76
The chronic psychological repercussions of NF1 have been the subject of two studies evaluating anxio-depressive disorders in adults 77, and children 78 : 19% of adults reported depressive symptoms and 15% anxiety disorders 77. In adults, the associations between the severity of anxio-depressive symptoms and the severity of the disease (calculated by a modified version of the Riccardi score 79) or the visibility of skin lesions (calculated according to the Ablon score 80) are significant (p<0.001) 77.
Sleep disturbances in patients with NF1 have also been evaluated in one study in adults 81 and another one in children 65The mean scores of ESS (Epworth Sleepiness Scale) 82 and PSQI (Pittsburgh Sleep Quality Index)) 83 which assess sleep disturbances, are abnormally high in these patients 81, with no correlation between the two scores.
In children with NF1, conduct problems (p £ 0.05), hyperactivity (p £ 0.01), emotional fragility (p £ 0.01) and total difficulty score (p £ 0.01) are significantly increased and correlated with sleep disturbances 65. These psychological and sleep impacts, associated with chronic pain, significantly contribute to the impairment of the quality of life of patients with NF1 84–86Adaptation of the quality of life (Skindex-16) and burden scores are underway for NF1: CutNF-Skindex and Burden of NF1 respectively.
The long-term management of these patients must integrate this dimension.
The risk of mortality is increased in patients with NF1. Compared to the general population, their life expectancy is shortened by 10 to 15 years 15,67,68,72,87–89. In France, the last NF1 mortality study dates from 2011 89and concerned data from 1985 patients between 1980 and 2005. All-cause mortality was significantly higher in the NF1 patient cohort, with a SMR (Standardized Mortality Ratio)of 2.02 (1.6-2.6) (p <0.0001). The main cause of death, available for 58 patients (86.6% of patients who died), was MPNST.
Main differential diagnosis
The main differential diagnosis of neurofibromatosis type 1 (NF1) is Legius syndrome. Like the diagnostic criteria for NF1, those for Legius syndrome are divided into two scenarios (A and B) depending on the absence (A) or presence (B) of an affected parent.
Thus, the diagnosis of Legius syndrome is made under the following conditions:
- The diagnostic criteria for Legius syndrome are met in an individual who does not have a parent diagnosed with Legius syndrome if the following criteria are present:
- Six or more "café-au-lait" macules bilaterally distributed and no other NF1-related diagnostic criteria except for axillaryor inguinal freckling.
- A heterozygous pathogenic SPRED1NF1 variant with a variant allele fraction of 50% in apparently normal tissue such as white blood cells.
- A child of a parent who meets the diagnostic criteria specified in A merits a diagnosis of NF1 if one or more of the criteria in A are present.
The presence of fewer than six café-au-lait macules does not exclude Legius syndrome.
Patient care
For the most symptomatic forms, NF1 requires multidisciplinary and multi-professional monitoring over the longterm, with specific research of the various above-mentioned complications and appropriate support, sometimes from an early age (with learning support, for instance).
This care is routinely organized near patients with reference to the appropriate referral centre of neurofibromatosis, when necessary.
The National Diagnostic and Care Protocols ("Protocoles Nationaux de Diagnostic et de Soin", PNDS) was updated in 2021, and it is available on the French High Authority of Health website or on the page of useful documents for healthcare professionals.
These complications are most often not specific to NF1, and their management does not differ from the same situations encountered in patients without NF1. However, several cases need to be clarified.
Pain is a very common symptom of NF1 and can accompany, or even reveal, many complications. The neuropathic component of pain is preponderant when it is secondary to compressive and/or transformed internal neurofibromas. The use of active treatments on neuropathic pain, in addition to conventional analgesics, is therefore essential for patients with NF1. These treatments include drugs from the family of antiepileptics and/or antidepressants.
For adult patients, many hospitals offer specialised pain management. Referral and competence centres most often work with a pain management team from the same hospital. For patients suffering from chronic pain in the Paris region, they can contact a specialised structure within the AP-HP, on this link..
The assessment and management of children's pain is complex and most often involves multidisciplinary and multi-professional teams. Various websites can help parents, but healthcare professionals may also contribute to the assessment and management of pain, such as:
Malignant peripheral nerve sheaths tumours (MPNSTs) are serious complications whose prognosis depends on the possibility of performing a broad surgical excision. After surgical treatment, when possible, adjuvant radiotherapy is most often offered. In case of a locally advanced and inoperable disease, or in the metastatic stage, poly-chemotherapy is proposed.
Cutaneous neurofibromas rarely evolve into MPNST, but they can be responsible for major aesthetic discomfort. The only treatments currently available are interventional treatments: CO2 laser2, conventional surgical excision, and electro-dissection. Aesthetic results depend on the quality of healing and the number of lesions to be treated.
Although the surgical treatment remains the first choice in the management of symptomatic plexiform neurofibromas, their partial or complete resection is not always feasible. In this context, several molecules have emerged, in particular MEK inhibitors. Multiple studies have demonstrated their efficacy as a treatment for symptomatic and inoperable internal neurofibromas.90–93. In 2021, selumetinib received marketing authorization at the European level for the following indication: inoperable plexiform neurofibromas in children. In 2023, its reimbursement was approved by social security. In 2025, selumetinib was recommended for approval in the European Union for the treatment of symptomatic and inoperable plexiform neurofibromas in adults with NF1. This recommendation by the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency is based on the results of the KOMET Phase III trial, a global Phase III, placebo-controlled, double-blind study conducted in adults with this condition. The trial demonstrated a statistically significant objective response rate of 20% in tumor size reduction.94In France, it is now possible to prescribe selumetinib as part of an early access program, prior to full marketing authorization, in adults. These treatments may be prescribed in specialized centers after multidisciplinary team discussions (RCP).
Bibliographic references
- Lammert M, Friedman JM, Kluwe L, Mautner VF. Prevalence of neurofibromatosis 1 in German children at elementary school enrollment. Arch Dermatol. 2005;141(1):71-74. doi:10.1001/archderm.141.1.71
- Evans DG, Howard E, Giblin C, et al. Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am J Med Genet A. 2010;152A(2):327-332. doi:10.1002/ajmg.a.33139
- Huson SM, Compston DA, Clark P, Harper PS. A genetic study of von Recklinghausen neurofibromatosis in south east Wales. I. Prevalence, fitness, mutation rate, and effect of parental transmission on severity. J Med Genet. 1989;26(11):704-711. doi:10.1136/jmg.26.11.704
- Kallionpää RA, Uusitalo E, Leppävirta J, Pöyhönen M, Peltonen S, Peltonen J. Prevalence of neurofibromatosis type 1 in the Finnish population. Genet Med Off J Am Coll Med Genet. 2018;20(9):1082-1086. doi:10.1038/gim.2017.215
- Huson SM, Compston DA, Harper PS. A genetic study of von Recklinghausen neurofibromatosis in south east Wales. II. Guidelines for genetic counselling. J Med Genet. 1989;26(11):712-721. doi:10.1136/jmg.26.11.712
- Poyhonen M, Kytölä S, Leisti J. Epidemiology of neurofibromatosis type 1 (NF1) in northern Finland. J Med Genet. 2000;37(8):632-636. doi:10.1136/jmg.37.8.632
- Uusitalo E, Leppävirta J, Koffert A, et al. Incidence and mortality of neurofibromatosis: a total population study in Finland. J Invest Dermatol. 2015;135(3):904-906. doi:10.1038/jid.2014.465
- DeBella K, Szudek J, Friedman JM. Use of the national institutes of health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics. 2000;105(3 Pt 1):608-614. doi:10.1542/peds.105.3.608
- Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 1988;45(5):575-578.
- Legius E, Messiaen L, Wolkenstein P, Pancza P, Avery RA, Do G. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: an international consensus recommendation. Genet Med. Published online May 19, 2021:23(8):1506-13. doi:10.1038/s41436-021-01170-5
- Bergqvist C, Servy A, Valeyrie-Allanore L, et al. Neurofibromatosis 1 French national guidelines based on an extensive literature review since 1966. Orphanet J Rare Dis. 2020;15(1):37. doi:10.1186/s13023-020-1310-3
- Duong TA, Bastuji-Garin S, Valeyrie-Allanore L, Sbidian E, Ferkal S, Wolkenstein P. Evolving pattern with age of cutaneous signs in neurofibromatosis type 1: a cross-sectional study of 728 patients. Dermatol Basel Switz. 2011;222(3):269-273. doi:10.1159/000327379
- Ortonne N, Wolkenstein P, Blakeley JO, et al. Cutaneous neurofibromas: Current clinical and pathologic issues. Neurology. 2018;91(2 Suppl 1):S5-S13. doi:10.1212/WNL.0000000000005792
- Darrigo LG, Geller M, Bonalumi Filho A, Azulay DR. Prevalence of plexiform neurofibroma in children and adolescents with type I neurofibromatosis. J Pediatr (Rio J). 2007;83(6):571-573. doi:10.2223/JPED.1718
- Uusitalo E, Rantanen M, Kallionpää RA, et al. Distinctive Cancer Associations in Patients With Neurofibromatosis Type 1. J Clin Oncol Off J Am Soc Clin Oncol. 2016;34(17):1978-1986. doi:10.1200/JCO.2015.65.3576
- Evans DGR, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39(5):311-314. doi:10.1136/jmg.39.5.311
- Ferner RE, Gutmann DH. International consensus statement on malignant peripheral nerve sheath tumors in neurofibromatosis. Cancer Res. 2002;62(5):1573-1577.
- Ducatman BS, Scheithauer BW, Piepgras DG, Reiman HM, Ilstrup DM. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer. 1986;57(10):2006-2021. doi:10.1002/1097-0142(19860515)57:10<2006::aid-cncr2820571022>3.0.co;2-6
- Evans DGR, Huson SM, Birch JM. Malignant peripheral nerve sheath tumours in inherited disease. Clin Sarcoma Res. 2012;2(1):17. doi:10.1186/2045-3329-2-17
- Tucker T, Wolkenstein P, Revuz J, Zeller J, Friedman JM. Association between benign and malignant peripheral nerve sheath tumors in NF1. Neurology. 2005;65(2):205-211. doi:10.1212/01.wnl.0000168830.79997.13
- Fertitta L, Jannic A, Zehou O, et al. Whole-body 18F-FDG-PET/MRI as a screening tool for the detection of malignant transformation in individuals with Neurofibromatosis type 1. J Invest Dermatol. Published online February 16, 2024:S0022-202X(24)00112-X. doi:10.1016/j.jid.2024.01.028
- Listernick R, Charrow J, Greenwald M, Mets M. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr. 1994;125(1):63-66. doi:10.1016/s0022-3476(94)70122-9
- Slamovits TL. Von Recklinghausen neurofibromatosis: II. Incidence of optic gliomata. Ophthalmology. 1985;92(5):714-715.
- Kornreich L, Blaser S, Schwarz M, et al. Optic pathway glioma: correlation of imaging findings with the presence of neurofibromatosis. AJNR Am J Neuroradiol. 2001;22(10):1963-1969.
- Deliganis AV, Geyer JR, Berger MS. Prognostic significance of type 1 neurofibromatosis (von Recklinghausen Disease) in childhood optic glioma. Neurosurgery. 1996;38(6):1114-1118; discussion 1118-1119. doi:10.1097/00006123-199606000-00010
- Listernick R, Darling C, Greenwald M, Strauss L, Charrow J. Optic pathway tumors in children: the effect of neurofibromatosis type 1 on clinical manifestations and natural history. J Pediatr. 1995;127(5):718-722. doi:10.1016/s0022-3476(95)70159-1
- Listernick R, Charrow J, Greenwald MJ, Esterly NB. Optic gliomas in children with neurofibromatosis type 1. J Pediatr. 1989;114(5):788-792. doi:10.1016/s0022-3476(89)80137-4
- Lewis RA, Gerson LP, Axelson KA, Riccardi VM, Whitford RP. von Recklinghausen neurofibromatosis. II. Incidence of optic gliomata. Ophthalmology. 1984;91(8):929-935. doi:10.1016/s0161-6420(84)34217-8
- Lund AM, Skovby F. Optic gliomas in children with neurofibromatosis type 1. Eur J Pediatr. 1991;150(12):835-838. doi:10.1007/BF01955002
- Prada CE, Hufnagel RB, Hummel TR, et al. The Use of Magnetic Resonance Imaging Screening for Optic Pathway Gliomas in Children with Neurofibromatosis Type 1. J Pediatr. 2015;167(4):851-856.e1. doi:10.1016/j.jpeds.2015.07.001
- Blazo MA, Lewis RA, Chintagumpala MM, Frazier M, McCluggage C, Plon SE. Outcomes of systematic screening for optic pathway tumors in children with Neurofibromatosis Type 1. Am J Med Genet A. 2004;127A(3):224-229. doi:10.1002/ajmg.a.20650
- Laue L, Comite F, Hench K, Loriaux DL, Cutler GB, Pescovitz OH. Precocious puberty associated with neurofibromatosis and optic gliomas. Treatment with luteinizing hormone releasing hormone analogue. Am J Dis Child 1960. 1985;139(11):1097-1100. doi:10.1001/archpedi.1985.02140130035025
- Boulanger JM, Larbrisseau A. Neurofibromatosis type 1 in a pediatric population: Ste-Justine’s experience. Can J Neurol Sci J Can Sci Neurol. 2005;32(2):225-231. doi:10.1017/s0317167100004017
- Pinson S, Créange A, Barbarot S, et al. [Recommendations for the treatment of neurofibromatosis type 1]. J Fr Ophtalmol. 2002;25(4):423-433.
- Akbarnia BA, Gabriel KR, Beckman E, Chalk D. Prevalence of scoliosis in neurofibromatosis. Spine. 1992;17(8 Suppl):S244-248. doi:10.1097/00007632-199208001-00005
- Ferner RE. Neurofibromatosis 1. Eur J Hum Genet EJHG. 2007;15(2):131-138. doi:10.1038/sj.ejhg.5201676
- Arrington DK, Danehy AR, Peleggi A, Proctor MR, Irons MB, Ullrich NJ. Calvarial defects and skeletal dysplasia in patients with neurofibromatosis Type 1. J Neurosurg Pediatr. 2013;11(4):410-416. doi:10.3171/2013.1.PEDS12409
- Ferner RE. Neurofibromatosis 1 and neurofibromatosis 2: a twenty first century perspective. Lancet Neurol. 2007;6(4):340-351. doi:10.1016/S1474-4422(07)70075-3
- Ramachandran M, Tsirikos AI, Lee J, Saifuddin A. Whole-spine magnetic resonance imaging in patients with neurofibromatosis type 1 and spinal deformity. J Spinal Disord Tech. 2004;17(6):483-491. doi:10.1097/01.bsd.0000133466.97241.50
- Brunetti-Pierri N, Doty SB, Hicks J, et al. Generalized metabolic bone disease in Neurofibromatosis type I. Mol Genet Metab. 2008;94(1):105-111. doi:10.1016/j.ymgme.2007.12.004
- Lammert M, Kappler M, Mautner VF, et al. Decreased bone mineral density in patients with neurofibromatosis 1. Osteoporos Int J Establ Result Coop Eur Found Osteoporos Natl Osteoporos Found USA. 2005;16(9):1161-1166. doi:10.1007/s00198-005-1940-2
- Vassilopoulou-Sellin R, Woods D, Quintos MT, Needle M, Klein MJ. Short stature in children and adults with neurofibromatosis. Pediatr Nurs. 1995;21(2):149-153.
- Virdis R, Sigorini M, Laiolo A, et al. Neurofibromatosis type 1 and precocious puberty. J Pediatr Endocrinol Metab JPEM. 2000;13 Suppl 1:841-844. doi:10.1515/jpem.2000.13.s1.841
- Lama G, Graziano L, Calabrese E, et al. Blood pressure and cardiovascular involvement in children with neurofibromatosis type1. Pediatr Nephrol Berl Ger. 2004;19(4):413-418. doi:10.1007/s00467-003-1397-5
- Fossali E, Signorini E, Intermite RC, et al. Renovascular disease and hypertension in children with neurofibromatosis. Pediatr Nephrol Berl Ger. 2000;14(8-9):806-810. doi:10.1007/s004679900260
- Virdis R, Balestrazzi P, Zampolli M, Donadio A, Street M, Lorenzetti E. Hypertension in children with neurofibromatosis. J Hum Hypertens. 1994;8(5):395-397.
- Tedesco MA, Di Salvo G, Ratti G, et al. Arterial distensibility and ambulatory blood pressure monitoring in young patients with neurofibromatosis type 1. Am J Hypertens. 2001;14(6 Pt 1):559-566. doi:10.1016/s0895-7061(00)01303-0
- Friedman JM, Arbiser J, Epstein JA, et al. Cardiovascular disease in neurofibromatosis 1: report of the NF1 Cardiovascular Task Force. Genet Med Off J Am Coll Med Genet. 2002;4(3):105-111. doi:10.1097/00125817-200205000-00002
- Neiman HL, Mena E, Holt JF, Stern AM, Perry BL. Neurofibromatosis and congenital heart disease. Am J Roentgenol Radium Ther Nucl Med. 1974;122(1):146-149. doi:10.2214/ajr.122.1.146
- Holt JF. 1977 Edward B. D. Neuhauser lecture: neurofibromatosis in children. AJR Am J Roentgenol. 1978;130(4):615-639. doi:10.2214/ajr.130.4.615
- Carey JC, Laub JM, Hall BD. Penetrance and variability in neurofibromatosis: a genetic study of 60 families. Birth Defects Orig Artic Ser. 1979;15(5B):271-281.
- Schorry EK, Stowens DW, Crawford AH, Stowens PA, Schwartz WR, Dignan PS. Summary of patient data from a multidisciplinary neurofibromatosis clinic. Neurofibromatosis. 1989;2(2):129-134.
- Colley A, Donnai D, Evans DG. Neurofibromatosis/Noonan phenotype: a variable feature of type 1 neurofibromatosis. Clin Genet. 1996;49(2):59-64. doi:10.1111/j.1399-0004.1996.tb04328.x
- Tonsgard JH, Yelavarthi KK, Cushner S, Short MP, Lindgren V. Do NF1 gene deletions result in a characteristic phenotype? Am J Med Genet. 1997;73(1):80-86. doi:10.1002/(sici)1096-8628(19971128)73:1<80::aid-ajmg16>3.0.co;2-n
- Ademiluyi SA, Sowemimo GO, Oyeneyin JO. Surgical experience in the management of multiple neurofibromatosis in Nigerians. West Afr J Med. 1989;8(1):59-65.
- Rasko JE, North KN, Favaloro EJ, Grispo L, Berndt MC. Attenuated platelet sensitivity to collagen in patients with neurofibromatosis type 1. Br J Haematol. 1995;89(3):582-588. doi:10.1111/j.1365-2141.1995.tb08367.x
- Korf BR, Carrazana E, Holmes GL. Patterns of seizures observed in association with neurofibromatosis 1. Epilepsia. 1993;34(4):616-620. doi:10.1111/j.1528-1157.1993.tb00437.x
- Kulkantrakorn K, Geller TJ. Seizures in neurofibromatosis 1. Pediatr Neurol. 1998;19(5):347-350. doi:10.1016/s0887-8994(98)00075-7
- Holmes MD. The causes of epilepsy: common and uncommon causes in adults and children. Arch Neurol. 2012;69(9):1212-1213. doi:10.1001/archneurol.2012.1266
- North KN, Riccardi V, Samango-Sprouse C, et al. Cognitive function and academic performance in neurofibromatosis. 1: consensus statement from the NF1 Cognitive Disorders Task Force. Neurology. 1997;48(4):1121-1127. doi:10.1212/wnl.48.4.1121
- Descheemaeker MJ, Plasschaert E, Frijns JP, Legius E. Neuropsychological profile in adults with neurofibromatosis type 1 compared to a control group. J Intellect Disabil Res JIDR. 2013;57(9):874-886. doi:10.1111/j.1365-2788.2012.01648.x
- Ferner RE, Hughes RA, Weinman J. Intellectual impairment in neurofibromatosis 1. J Neurol Sci. 1996;138(1-2):125-133. doi:10.1016/0022-510x(96)00022-6
- Acosta MT, Gioia GA, Silva AJ. Neurofibromatosis type 1: new insights into neurocognitive issues. Curr Neurol Neurosci Rep. 2006;6(2):136-143. doi:10.1007/s11910-996-0036-5
- Hyman SL, Arthur Shores E, North KN. Learning disabilities in children with neurofibromatosis type 1: subtypes, cognitive profile, and attention-deficit-hyperactivity disorder. Dev Med Child Neurol. 2006;48(12):973-977. doi:10.1017/S0012162206002131
- Johnson H, Wiggs L, Stores G, Huson SM. Psychological disturbance and sleep disorders in children with neurofibromatosis type 1. Dev Med Child Neurol. 2005;47(4):237-242. doi:10.1017/s0012162205000460
- Seminog OO, Goldacre MJ. Risk of benign tumours of nervous system, and of malignant neoplasms, in people with neurofibromatosis: population-based record-linkage study. Br J Cancer. 2013;108(1):193-198. doi:10.1038/bjc.2012.535
- Madanikia SA, Bergner A, Ye X, Blakeley JO. Increased risk of breast cancer in women with NF1. Am J Med Genet A. 2012;158A(12):3056-3060. doi:10.1002/ajmg.a.35550
- Walker L, Thompson D, Easton D, et al. A prospective study of neurofibromatosis type 1 cancer incidence in the UK. Br J Cancer. 2006;95(2):233-238. doi:10.1038/sj.bjc.6603227
- Wang X, Levin AM, Smolinski SE, Vigneau FD, Levin NK, Tainsky MA. Breast cancer and other neoplasms in women with neurofibromatosis type 1: a retrospective review of cases in the Detroit metropolitan area. Am J Med Genet A. 2012;158A(12):3061-3064. doi:10.1002/ajmg.a.35560
- Sharif S, Moran A, Huson SM, et al. Women with neurofibromatosis 1 are at a moderately increased risk of developing breast cancer and should be considered for early screening. J Med Genet. 2007;44(8):481-484. doi:10.1136/jmg.2007.049346
- Uusitalo E, Kallionpää RA, Kurki S, et al. Breast cancer in neurofibromatosis type 1: overrepresentation of unfavourable prognostic factors. Br J Cancer. 2017;116(2):211-217. doi:10.1038/bjc.2016.403
- Evans DGR, O’Hara C, Wilding A, et al. Mortality in neurofibromatosis 1: in North West England: an assessment of actuarial survival in a region of the UK since 1989. Eur J Hum Genet EJHG. 2011;19(11):1187-1191. doi:10.1038/ejhg.2011.113
- Daly MB, Pilarski R, Berry M, et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast and Ovarian, Version 2.2017. J Natl Compr Cancer Netw JNCCN. 2017;15(1):9-20. doi:10.6004/jnccn.2017.0003
- Guillamo JS, Créange A, Kalifa C, et al. Prognostic factors of CNS tumours in Neurofibromatosis 1 (NF1): a retrospective study of 104 patients. Brain J Neurol. 2003;126(Pt 1):152-160. doi:10.1093/brain/awg016
- Salvi PF, Lorenzon L, Caterino S, Antolino L, Antonelli MS, Balducci G. Gastrointestinal stromal tumors associated with neurofibromatosis 1: a single centre experience and systematic review of the literature including 252 cases. Int J Surg Oncol. 2013;2013:398570. doi:10.1155/2013/398570
- Miettinen M, Fetsch JF, Sobin LH, Lasota J. Gastrointestinal stromal tumors in patients with neurofibromatosis 1: a clinicopathologic and molecular genetic study of 45 cases. Am J Surg Pathol. 2006;30(1):90-96. doi:10.1097/01.pas.0000176433.81079.bd
- Doser K, Andersen EW, Kenborg L, et al. Clinical characteristics and quality of life, depression, and anxiety in adults with neurofibromatosis type 1: A nationwide study. Am J Med Genet A. 2020;182(7):1704-1715. doi:10.1002/ajmg.a.61627
- Pasini A, Lo-Castro A, Di Carlo L, et al. Detecting anxiety symptoms in children and youths with neurofibromatosis type I. Am J Med Genet Part B Neuropsychiatr Genet Off Publ Int Soc Psychiatr Genet. 2012;159B(7):869-873. doi:10.1002/ajmg.b.32095
- Barton B, North K. The self-concept of children and adolescents with neurofibromatosis type 1. Child Care Health Dev. 2007;33(4):401-408. doi:10.1111/j.1365-2214.2006.00717.x
- Ablon J. Gender response to neurofibromatosis 1. Soc Sci Med 1982. 1996;42(1):99-109. doi:10.1016/0277-9536(95)00076-3
- Leschziner GD, Golding JF, Ferner RE. Sleep disturbance as part of the neurofibromatosis type 1 phenotype in adults. Am J Med Genet A. 2013;161A(6):1319-1322. doi:10.1002/ajmg.a.35915
- Johns MW. A new method for measuring daytime sleepiness: the Epworth sleepiness scale. Sleep. 1991;14(6):540-545. doi:10.1093/sleep/14.6.540
- Buysse DJ, Reynolds CF, Monk TH, Berman SR, Kupfer DJ. The Pittsburgh Sleep Quality Index: a new instrument for psychiatric practice and research. Psychiatry Res. 1989;28(2):193-213. doi:10.1016/0165-1781(89)90047-4
- Vranceanu AM, Merker VL, Park E, Plotkin SR. Quality of life among adult patients with neurofibromatosis 1, neurofibromatosis 2 and schwannomatosis: a systematic review of the literature. J Neurooncol. 2013;114(3):257-262. doi:10.1007/s11060-013-1195-2
- Vranceanu AM, Merker VL, Park ER, Plotkin SR. Quality of life among children and adolescents with neurofibromatosis 1: a systematic review of the literature. J Neurooncol. 2015;122(2):219-228. doi:10.1007/s11060-015-1725-1
- Garwood MM, Bernacki JM, Fine KM, Hainsworth KR, Davies WH, Klein-Tasman BP. Physical, cognitive, and psychosocial predictors of functional disability and health-related quality of life in adolescents with neurofibromatosis-1. Pain Res Treat. 2012;2012:975364. doi:10.1155/2012/975364
- Rasmussen SA, Yang Q, Friedman JM. Mortality in neurofibromatosis 1: an analysis using U.S. death certificates. Am J Hum Genet. 2001;68(5):1110-1118. doi:10.1086/320121
- Zöller M, Rembeck B, Akesson HO, Angervall L. Life expectancy, mortality and prognostic factors in neurofibromatosis type 1. A twelve-year follow-up of an epidemiological study in Göteborg, Sweden. Acta Derm Venereol. 1995;75(2):136-140. doi:10.2340/0001555575136140
- Duong TA, Sbidian E, Valeyrie-Allanore L, et al. Mortality associated with neurofibromatosis 1: a cohort study of 1895 patients in 1980-2006 in France. Orphanet J Rare Dis. 2011;6:18. doi:10.1186/1750-1172-6-18
- Dombi E, Baldwin A, Marcus LJ, et al. Activity of Selumetinib in Neurofibromatosis Type 1–Related Plexiform Neurofibromas. https://doi.org/10.1056/NEJMoa1605943. doi:10.1056/NEJMoa1605943
- Weiss BD, Wolters PL, Plotkin SR, et al. NF106: A Neurofibromatosis Clinical Trials Consortium Phase II Trial of the MEK Inhibitor Mirdametinib (PD-0325901) in Adolescents and Adults With NF1-Related Plexiform Neurofibromas. J Clin Oncol Off J Am Soc Clin Oncol. 2021;39(7):797-806. doi:10.1200/JCO.20.02220
- Gross AM, Wolters PL, Dombi E, et al. Selumetinib in Children with Inoperable Plexiform Neurofibromas. N Engl J Med. 2020;382(15):1430-1442. doi:10.1056/NEJMoa1912735
- Fisher MJ, Shih CS, Rhodes SD, et al. Cabozantinib for neurofibromatosis type 1-related plexiform neurofibromas: a phase 2 trial. Nat Med. 2021;27(1):165-173. doi:10.1038/s41591-020-01193-6
- Chen AP, Coyne GO, Wolters PL, et al. Efficacy and safety of selumetinib in adults with neurofibromatosis type 1 and symptomatic, inoperable plexiform neurofibromas (KOMET): a multicentre, international, randomised, placebo-controlled, parallel, double-blind, phase 3 study. Lancet Lond Engl. 2025;405(10496):2217-2230. doi:10.1016/S0140-6736(25)00986-9